3. Basic usage
This chapter will introduce some basic usage of DMFF. All scripts can be found in examples/ directory in which Jupyter notebook-based demos are provided.
3.1 Compute energy
DMFF uses OpenMM to parse input files, including coordinates file, topology specification file and force field parameter file. Then, the core class Hamiltonian inherited from openmm.ForceField will be initialized and the method createPotential will be called to create differentiable potential energy functions for different energy terms. Take parametrzing an organic moleclue with GAFF2 force field as an example:
import jax
import jax.numpy as jnp
import openmm.app as app
import openmm.unit as unit
from dmff import Hamiltonian, NeighborList
app.Topology.loadBondDefinitions("lig-top.xml")
pdb = app.PDBFile("lig.pdb")
ff = Hamiltonian("gaff-2.11.xml", "lig-prm.xml")
potentials = ff.createPotential(pdb.topology)
for k in potentials.dmff_potentials.keys():
pot = potentials.dmff_potentials[k]
print(pot)
In this example, lig.pdb is the PDB file containing atomic coordinates, and lig-top.xml specifying bond connections within a molecule and this information is required by openmm.app to generate molecular topology. Note that this file is not always required, if bond conncections are defined in .pdb file by CONNECT keyword. gaff-2.11.xml contains GAFF2 force field parameters (bonds, angles, torsion and vdW), and lig-prm.xml contains atomic partial charges (GAFF2 requests a user-defined charge assignment process). This xml format is compatitable with OpenMM definitions, and a detailed description can be found in OpenMM user guide or XML-format force fields section.
If you run this script in examples/classical, you will get the following output.
<function HarmonicBondJaxGenerator.createForce.<locals>.potential_fn at 0x112504af0>
<function HarmonicAngleJaxGenerator.createForce.<locals>.potential_fn at 0x1124cd820>
<function PeriodicTorsionJaxGenerator.createForce.<locals>.potential_fn at 0x18509b790>
<function NonbondJaxGenerator.createForce.<locals>.potential_fn at 0x18509baf0>
The force field parameters are stored as a Python dict in the Hamiltonian.
params = ff.getParameters()
nbparam = params['NonbondedForce']
nbparam
{
'sigma': DeviceArray([0.33152124, ...], dtype=float32),
'epsilon': DeviceArray([0.4133792, ...], dtype=float32),
'epsfix': DeviceArray([], dtype=float32),
'sigfix': DeviceArray([], dtype=float32),
'charge': DeviceArray([-0.75401515, ...], dtype=float32),
'coulomb14scale': DeviceArray([0.8333333], dtype=float32),
'lj14scale': DeviceArray([0.5], dtype=float32)
}
Each generated function will read coordinates, box, pairs and force field parameters as inputs.
positions = jnp.array(pdb.getPositions(asNumpy=True).value_in_unit(unit.nanometer))
box = jnp.array([
[10.0, 0.0, 0.0],
[ 0.0, 10.0, 0.0],
[ 0.0, 0.0, 10.0]
])
nbList = NeighborList(box, rc=4)
nbList.allocate(positions)
pairs = nbList.pairs
Note that in order to take advantages of the auto-differentiable implementation in JAX, the input arrays have to be jax.numpy.ndarray, otherwise DMFF will raise an error. pairs is a \(N\times 2\) integer array in which each row specifying atoms condsidered as neighbors within rcut. As shown above, this can be calculated with dmff.NeighborList class which is supported by jax_md.
The potential energy function will give energy (a scalar, in kJ/mol) as output:
nbfunc = potentials.dmff_potentials['NonbondedForce']
nbene = nbfunc(positions, box, pairs, params)
print(nbene)
If everything works fine, you will get -425.41412 as a result. In addition, you can also use getPotentialFunc() and getParameters() to obtain the whole potential energy function and force field parameter set, instead of seperated functions for different energy terms.
efunc = potentials.getPotentialFunc()
params = ff.getParameters()
totene = efunc(positions, box, pairs, params)
3.2 Compute forces
Different from conventional programming frameworks, explicit definition of atomic force calculation functions are no longer needed. Instead, the forces can be evaluated in an automatic manner with jax.grad.
pos_grad_func = jax.grad(efunc, argnums=0)
force = -pos_grad_func(positions, box, pairs, params)
3.3 Compute parametric gradients
Similarly, the derivative of energy with regard to force field parameters can also be computed easily.
param_grad_func = jax.grad(nbfunc, argnums=-1)
pgrad = param_grad_func(positions, box, pairs, params)
print(pgrad["NonbondedForce"]["charge"])
[ 652.7753 55.108738 729.36115 -171.4929 502.70837
-44.917206 129.63994 -142.31796 -149.62088 453.21503
46.372574 140.15303 575.488 461.46902 294.4358
335.25153 27.828705 671.3637 390.8903 519.6835
220.51129 238.7695 229.97302 210.58838 231.8734
196.40994 237.08563 35.663574 457.76416 77.4798
256.54382 402.2121 611.9573 440.8465 -52.09662
421.86688 592.46265 237.98883 110.286194 150.65375
218.61087 240.20477 -211.85376 150.7331 310.89404
208.65228 -139.23026 -168.8883 114.3645 3.7261353
399.6282 298.28455 422.06445 526.18463 521.27563
575.85767 606.74744 394.40845 549.84033 556.4724
485.1427 512.1267 558.55896 560.4667 562.812
333.74194 ]
3.4 Parametrize molecules with SMIRKS-based force field
3.4.1 Background
Besides atom-typing based methods, DMFF also supports assigning force field parameters with SMIRKS. SMIRKS is an extenstion of SMARTS language which allows users not only to specify chemical substructures with certain patterns, but also to numerically tag the matched atoms for assigning parameters. This approach avoid the duplicate atom-typing definition process, which enables new parameters to be easily introduced to existing force field parameters sets. OpenFF[1-2] series are examples of SMIRKS-based force fields for organic molecules.
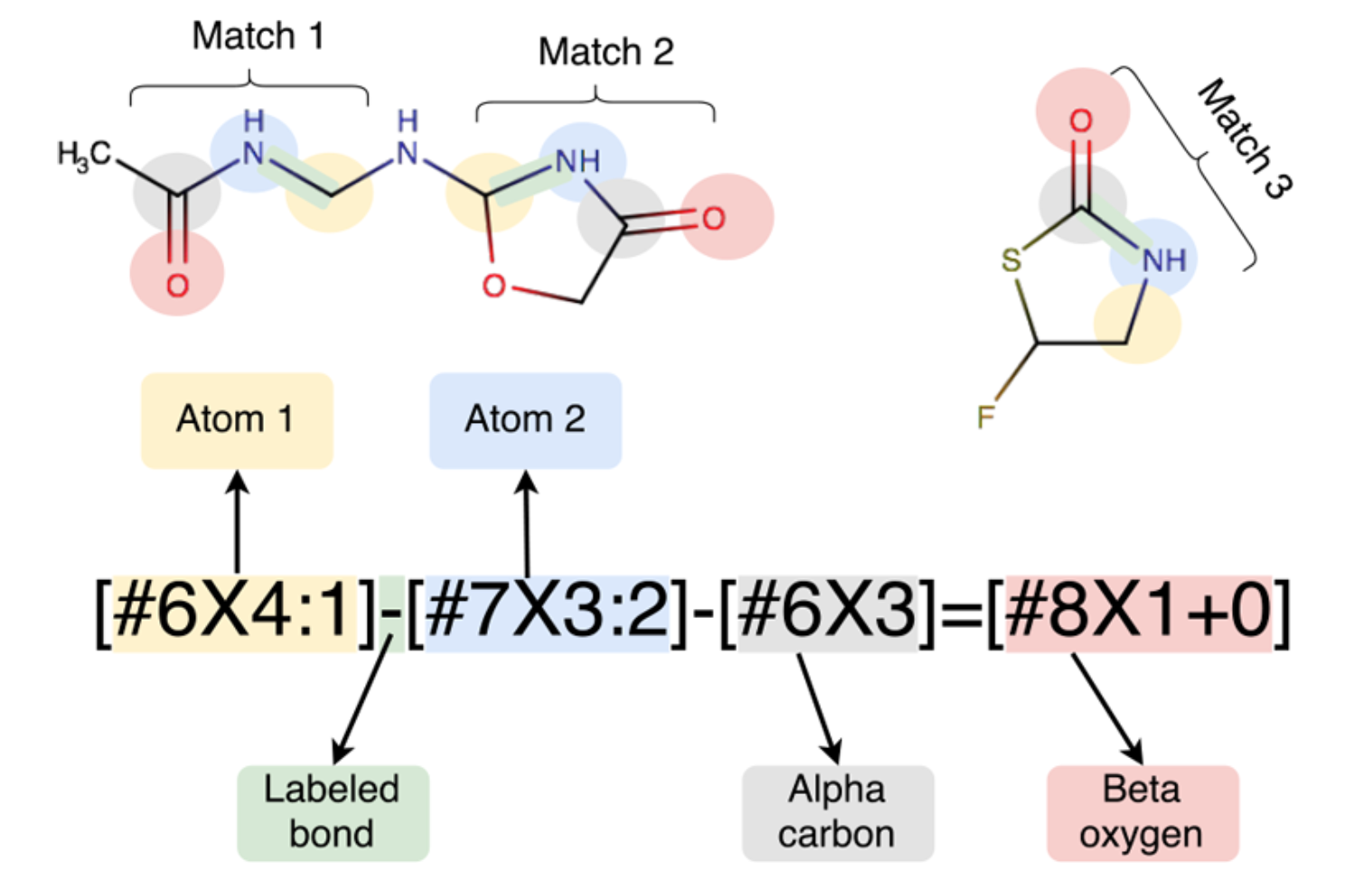
3.4.2 Parametrize molecules with SMIRKS by DMFF
The SMIRKS pattern matching is supported by RDKit package, which can be install with conda:
conda install rdkit -c conda-forge
To begin with, we need a molecule encoded in rdkit.Chem.Mol object. As an example, we will load a N-methylacetamide molecule defined in examples/smirks/C3H7NO.mol:
from rdkit import Chem
from dmff import Hamiltonian
mol = Chem.MolFromMolFile("C3H7NO.mol", removeHs=False) # hydrogens must be preserved
Then load force field parameters in xml format. Instuctions about how to write a SMIRKS-based force field XML file can be found in the Chapter 4.
h_smk = Hamiltonian("C3H7NO.xml", noOmmSys=True)
Note that the argument noOmmSys is set to False so that DMFF will not create an openmm system, as openmm does not support SMIRKS-based force field definitions.
Build an openmm topology and parametrize the molecule to create differentiable potential energy functions:
top = h_smk.buildTopologyFromMol(mol)
potObj = h_smk.createPotential(top, rdmol=mol)
So far, we can utilize this dmff.Potential object to calculate energy and forces as we did in the previous sections.
3.4.3 Bond Charge Correction (BCC) and Virtual Sites
Bond charge correction[3-4] is an approach to obtain high-accuracy atomic partial charges (e.g. HF/6-31G* ESP-fit charges) by adopting corrections to low-accuracy atomic charges (e.g. AMI Mulliken charges). In order to ensure a zero of total correction values within a molecule, these correction parameters are usually defined based on bond types, which suggests that they can also be defined by SMIRKS patterns.
Virtual sites are additional off-centered charged sites which are introduced to improve the desciption of electrostatic effects caused by sigma hole (halogen bond) or lone pairs. The positions of virtual sites are calculated directly by its parent atoms, not by integrating the equations of motion. This approach is well known for its application in TIP4P[5] and TIP5P[6] water models, and it also proves to be useful in drug-like moelcular force fields like OPLS series[7-8]. Basically, the parameters to define a virtual site includes : where to add virtual sites, how the virtual sites' positions are determined and the charges.
Not surprisingly, all these parameters can all be defined in SMIRKS pattern and as well as can be parsed with DMFF by adding terms in <NonbondedForce>, such as:
<NonbondedForce>
<BondChargeCorrection smirks="[#0:1]~[#17:2]" bcc="0.000000"/>
<BondChargeCorrection smirks="[#0:1]~[#35:2]" bcc="0.000000"/>
<BondChargeCorrection smirks="[#0:1]~[#53:2]" bcc="0.000000"/>
<BondChargeCorrection smirks="[#0:1]~[#7:2]" bcc="0.000000"/>
<VirtualSite smirks="[#17,#35:1]-[#6X3;a:2]" vtype="1" distance="0.1600" />
<VirtualSite smirks="[#53:1]-[#6X3;a:2]" vtype="1" distance="0.1800" />
<VirtualSite smirks="[#7X2;a:1](:[*;a:2]):[*;a:3]" vtype="2" distance="0.0400" />
</NonbondedForce>
In Chapter 4, we will explain the meaning of these XML-format parameters in detail. Here, we will give a simple example to parametrize 2-chloropyridine with BCC parameters and virtual sites.
As introduced above, we first load molecule and force field parameters.
import jax.numpy as jnp
from rdkit import Chem
from dmff import Hamiltonian
mol = Chem.MolFromMolFile("clpy.mol", removeHs=False)
h_vsite = Hamiltonian("clpy_vsite.xml", noOmmSys=True)
Next, we build the dmff potential. We can see the BCC and virtual site parameters are successfully parsed.
top = h_vsite.buildTopologyFromMol(mol)
potObj = h_vsite.createPotential(top, rdmol=mol)
Then we can add virtual sites to the molecule and obtain a new rdkit.Chem.Mol object.
mol_vsite = h_vsite.addVirtualSiteToMol(rdmol, h_vsite.getParameters())
Chem.MolToMolFile(mol_vsite, "clpy_vsite.mol")
By dumping this molecule to mol file and visualize it, we can see that as expected, two virtual sites are added along the bond between aromatic carbon (arC) and chloroine and also along the bisector of the arC-N-arC angle.
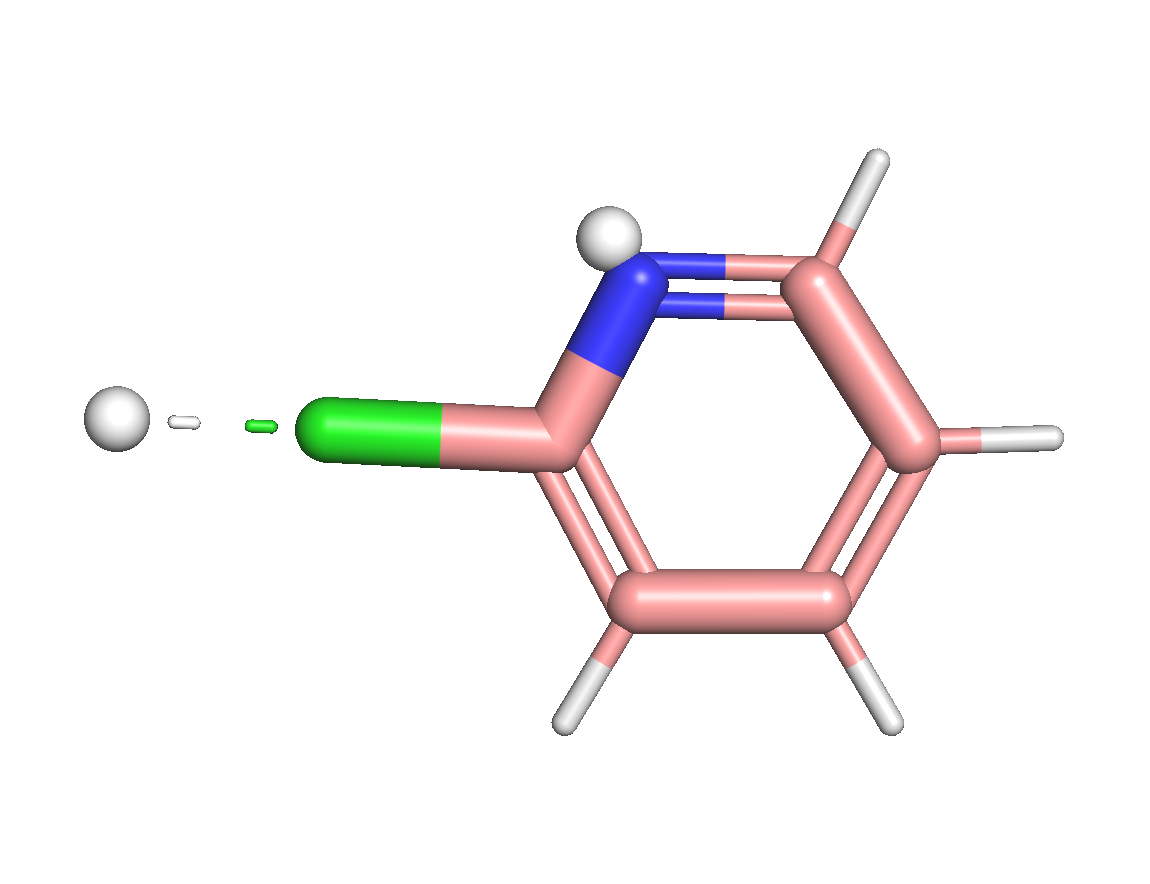
Alternatively, we can also add coordinates of virtual sites by taking atomic positions matrix as an input.
pos = jnp.array(mol.GetConformer().GetPositions()) / 10 # convert angstrom to nm
pos_vsite = h_vsite.addVirtualSiteCoords(pos, h_vsite.getParameters())